Les femmes et les hommes peuvent-ils vivre sans estrogènes ? – le foie impliqué en reproduction – AMH et grossesse chez les lupiques – Infertilité masculine,histoire de goût ? – cause immunologique d’insuffisance lactotrope isolée – dilatation aortique et syndrome de Turner
Les femmes et les hommes peuvent-ils vivre sans estrogènes ?
Sophie Christin-Maitre (Paris)
Le premier homme avec une perte de fonction du récepteur des estrogènes, ER?, a été décrit à Chicago il y a 19 ans (1). Ce patient avait consulté car il continuait de grandir alors qu’il avait 26 ans. Son âge osseux était de 15 ans et il présentait une ostéoporose majeure. L’été dernier, l’équipe de L Layman, aux Etats-Unis, a décrit la première femme sans estrogènes (2). Cette patiente, âgée de 18 ans, a consulté pour une aménorrhée primaire, une absence de développement mammaire et quelques douleurs pelviennes. Son IMC était de 16. Le bilan hormonal a montré un taux plasmatique d’estradiol très élevé à 3 500 pg/ml (N : 11 à 210) et des gonadotrophines non freinées, LH = 9,6 UI/L, FSH = 6,7 UI/L. Son âge osseux était de 13,5 ans et sa masse osseuse était très diminuée. Une IRM hypophysaire a éliminé un adénome gonadotrope. L’échographie pelvienne a montré des ovaires multikystiques. L’hypothèse d’une mutation « perte de fonction » du récepteur ER? a été évoquée. La patiente présente une mutation homozygote du gène codant pour le récepteur ER?, située dans la zone de liaison du ligand, dans une région très conservée à travers les espèces. L’expression in vitro du récepteur muté a confirmé une perte de fonction de ce récepteur par rapport au récepteur sauvage. Un traitement par estrogènes oraux a été tenté pendant 5 mois, il a été inefficace sur le volume mammaire. Un traitement par progestatif a réussi par son effet antigonadotrope à diminuer le taux d’estradiol plasmatique d’E2 et le volume des kystes ovariens. Les gonadotrophines n’étaient cependant pas très élevées, probablement en raison d’une sécrétion d’inhibine A par la patiente. Ce cas de syndrome de résistance aux estrogènes, médié par le récepteur ER?, illustre le rôle de l’E2 chez la femme sur la glande mammaire, sur l’os et sur l’axe hypothalamo-hypophysaire.
Et les hommes sans estrogènes ? Joel Finkelstein et son équipe viennent de rapporter dans le NEJM, une étude réalisée chez des hommes normaux volontaires, traités initialement par agonistes de la GnRH puis répartis par tirage au sort entre des traitements de placebo ou des doses croissantes de testostérone en gel (3). Une partie de la cohorte était de plus traitée par un inhibiteur de l’aromatase pour inhiber la synthèse d’estradiol. Cette étude montre que la composition corporelle est modifiée par les androgènes, mais aussi par les estrogènes. En effet, alors que l’étude n’a duré que 18 semaines, le manque d’estradiol a induit une augmentation de la masse grasse. Les hommes sous anastrozole ont présenté de plus une diminution de la fonction sexuelle, ce qui démontre que les estrogènes agissent en synergie avec les androgènes sur la fonction sexuelle.
En conclusion, les études de l’été dernier confirment le fait que les estrogènes sont très importants, aussi bien chez l’homme que chez la femme. Ils jouent un rôle très important sur la masse osseuse et la répartition de la masse grasse. Les effets de l’E2 sur l’insulinorésistance nécessitent d’être précisés car aucune anomalie de la régulation de la glycémie n’a été mise en évidence chez la jeune femme avec une résistance aux estrogènes.
Références bibliographiques
1. Smith EP, Boyd J, Frank GR, et al. Estrogen resistance caused by a mutation in the estrogen-receptor gene in a man.
N Engl J Med 1994;331:1056-61.
2. Quaynor SD, Stradtman EW Jr, Kim HG et al. Delayed puberty and estrogen resistance in a woman with estrogen receptor ?
variant. N Engl J Med. 2013 Jul 11;369:164-71
3. Finkelstein J Lee H, Burnett-Bowie SA et al. Gonadal steroids and body composition N Engl J Med 2013;369:1011-22
Nouvelle importante : le foie est un organe impliqué en reproduction, par l’intermédiaire du FGF21 ou Fibroblast growth factor21 !
Bruno Donadille (Paris)
Les liens entre l’état nutritionnel et la reproduction des mammifères sont établis depuis de nombreuses années. Il est connu que la dénutrition inhibe la reproduction, mais les liens restaient mal connus. Une équipe américaine vient de démontrer le rôle d’une protéine, FGF21, dans la régulation de l’axe gonadotrope (1). Leurs expériences ont utilisé un modèle de souris transgéniques, qui surexpriment cette protéine de 209 AA, clonée en 2000 par Nishimura et al. Membre de la superfamille des FGFs, FGF21 est synthétisée au niveau du foie et du tissu adipeux à partir d’un gène situé sur le chromosome 19. Cette protéine présente 75 % d’homologie entre la souris et l’homme (OMIM 603891). Sa signalisation passe par un corécepteur Klotho et la voie des ras/MAP kinases, sous contrôle transcriptionnel hépatique par le système PPAR. En physiologie, les taux plasmatiques de FGF21 produits par le foie augmentent pendant le jeûne. De plus, il a été montré qu’elle passe la barrière hémato-encéphalique.
Les souris femelles qui surexpriment FGF21 (TgFGF21) sont infertiles. Elles ont une puberté retardée, une anovulation avec une LH significativement basse par rapport aux souris témoins. Après ovariectomie, ces souris TgFGF21 ont, malgré l’administration exogène d’œstradiol, un pic de LH peu élevé, ce qui suggère qu’elles présentent un déficit hypothalamo-hypophysaire. Les auteurs ont montré que ce déficit est d’origine hypothalamique car le pic de LH est restauré après administration de la GnRH.
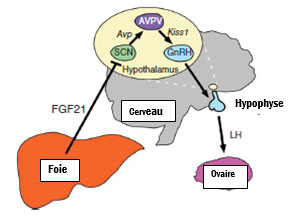
En physiologie, les neurones vasopressinergiques supra-thalamiques sont situés en amont du système neuronal Kiss situés dans les noyaux arqués (feedback négatif de l’estradiol) et de la région périventriculaire (AVPV) (feedback positif de l’estradiol). Les neurones Kiss sont en relation avec les neurones à GnRH et modulent la sécrétion des gonadotrophines hypophysaires qui induisent l’ovulation. Les souris femelles TgFGF21 ont une expression diminuée d’AVP dans les neurones vasopressinergiques et une diminution de Kiss1 dans le noyau périventriculaire mais non dans le noyau arqué. Le pic de LH est restauré chez les souris TgFGF21 après un traitement par AVP et/ou Kisspeptine. Ces expériences démontrent que FGF21 joue un rôle important sur les neurones Kiss situés dans le noyau périventriculaire.
A l’inverse des souris femelles, les souris mâles TgFGF21 sont fertiles. Cette différence dans l’axe gonadotrope pourrait être due à l’absence physiologique d’expression des neurones Kiss dans l’AVPV chez les mâles.
Grâce à la technique Cre/Lox, les auteurs ont généré des souris transgéniques, invalidées pour le co-recepteur Klotho, directement dans la région cérébrale antérieure. Ces souris ont une fertilité perturbée, ce qui illustre le rôle de ce co-recepteur dans l’axe gonadotrope. Après perfusion continue de FGF21 aux mêmes taux qu’en période de jeûne et sans modification du poids ou de la leptine, une anovulation est obtenue.
Si le tissu adipeux était connu comme jouant un rôle important en reproduction par l’intermédiaire de la leptine, cet article est la première illustration du rôle du foie dans la régulation de l’axe gonadotrope. Il reste à démontrer que cette nouvelle voie hépato-neuroendocrine contribue à l’anovulation en période de dénutrition dans l’espèce humaine.
Référence bibliographique
1. Owen BM, Bookout A, Ding X et al. FGF21 contributes to neuroendocrine control of female reproduction. Nature Medicine 2013;19:1153-1156
Figure : Modèle des liens entre le foie et l’axe gonadotrope (SCN: système nerveux central ; Avp : vasopressine, AVPV : noyau périventriculaire, Kiss 1 : neurones Kiss, GnRH Gonadotropin Releasing Hormone, FGF21 (Fibroblast Growth Factor 21)
Niveaux d’AMH et grossesse chez les patientes lupiques traitées ou non par cyclophosphamide
Anne Bachelot (Paris)
Le cyclophosphamide est un des traitements de référence du lupus systémique, notamment en cas d’atteinte rénale. Il peut être à l’origine d’insuffisance ovarienne prématurée. L’AMH a été mesurée chez 56 patientes lupiques de moins de 40 ans ayant été traitées par cyclophosphamide et chez 56 patientes lupiques non exposées au cyclophosphamide appariées sur l’âge, sur les tubes de sérothèque de l’étude PLUS. L’ensemble des patientes ont été interrogées en mai 2012 sur leur projet parental. L’âge moyen des 112 patientes était de 31,6 ± 5,8 ans. Les taux d’AMH étaient bas (1,21 ± 1,01 ng/mL) et étaient significativement plus bas chez les patientes exposées au cyclophosphamide (p = 0,03) et chez les patientes âgées de plus de 30 ans (p = 0,02). Sur une période de suivi de 4,2 [2,5-4,8] ans, 38 patientes ont eu un projet parental et 32 ont eu une grossesse (84,2 %). En analyse univariée, le risque d’échec du projet parental était associé à la dose cumulée de cyclophosphamide (p = 0,007) et à l’âge (p = 0,02), mais pas au taux d’AMH. Cette étude confirme donc que les taux d’AMH sont bas chez les patientes lupiques, même en l’absence d’exposition au cyclophosphamide. Les mécanismes à l’origine de cette observation sont encore méconnus (réduction de la réserve ovarienne chez les patientes lupiques, interférence d’anticorps avec le dosage ?). Néanmoins, les taux de grossesse sont satisfaisants (84,2 %), les échecs étant associés à l’exposition au cyclophosphamide, mais pas aux taux d’AMH.
Référence bibliographique
Morel N, Bachelot A, Chakhtoura H et al. Study of Anti-Müllerian Hormone and its relation to the subsequent probability of pregnancy in 112 patients with systemic lupus erythematosus, exposed or not to cyclophosphamide J Clin Endocrinol Metab 2013;98:3785–3792
Infertilité masculine, une histoire de goût ?
Léopoldine Bricaire (Paris)
À ce jour, dans 30 à 50 % des cas d’infertilité masculine, il existe une oligospermie ou azoospermie sans anomalie hormonale ou étiologie connue d’infertilité. Les récepteurs du goût TAS1R (Type 1 Taste Receptors) sont des récepteurs couplés à la protéine G hétéro-trimérique nommée gustducine dont les ligands sont les sucres, la saccharine ou des acides aminés modulateurs du goût. Ils sont exprimés dans les papilles, le tractus gastro-intestinal mais également en grande quantité dans le testicule et le sperme, notamment pour le sous-type TAS1R3. L’hypothèse d’une cause environnementale d’infertilité via l’action de ces récepteurs a été testée par l’équipe new-yorkaise de Margolskee dans un modèle murin.
Cette équipe a montré que des souris mâles invalidées à la fois pour TAS1R3 et pour la sous-unité ? de la gustducine (GNAT3) présentent une infertilité avec une altération de la spermatogénèse. Ils ont de plus généré un modèle de souris invalidées pour TAS1R3 et GNAT3 mais qui expriment le récepteur humain TAS1R3 qui est susceptible d’être inhibé par les fibrates ou les phénoxy-herbicides. Le blocage du récepteur TAS1R3 humain induit par un traitement par clofibrate chez ces souris aboutit rapidement à une infertilité masculine avec des anomalies de la spermatogénèse (malformations et immobilité du sperme), rapidement réversibles à l’arrêt du traitement. Des études complémentaires sur les fonctions de ces récepteurs dans le testicule sont nécessaires mais ceci suggère un effet potentiellement délétère des composés phénoxy, notamment certains agents environnementaux, via ce récepteur sur la fonction reproductrice masculine. Ils pourraient être impliqués dans les phénomènes d’oligo- ou azoospermies inexpliquées. Le développement d’activateurs de TAS1R3 ou GNAT3 pourraient par ailleurs constituer un traitement potentiel des infertilités masculines liées à des anomalies de ces voies de signalisation.
Référence bibliographique
Mosinger B, Redding KM, Parker MR et al. PNAS 2013; 110:12319-24
Une cause immunologique prouvée d’insuffisance lactotrope isolée… et une nouvelle étiologie d’hyperprolactinémie
Léopoldine Bricaire (Paris)
Le déficit en prolactine s’intègre le plus souvent dans le cadre d’un déficit anté-hypophysaire combiné – qu’il s’agisse d’une cause vasculaire (syndrome de Sheehan), d’une tumeur hypothalamo-hypophysaire, d’une maladie inflammatoire chronique ou infectieuse – ou encore dans le cadre de causes génétiques (défaut de protéine G, mutations de facteurs de transcription tels que POU1F1, PROP1, LHX3, LHX4, HESX1, OTX2, LSD1). Une insuffisance lactotrope isolée est extrêmement rare. Elle est diagnostiquée lors de l’absence de montée laiteuse en post-partum.
L’équipe américaine de Baltimore a décrit l’observation originale d’une patiente de 39 ans, sans antécédent particulier, en dehors d’une thyroïdite de Hashimoto à l’âge de 33 ans, n’ayant pas eu de montée laiteuse après 2 grossesses de déroulement normal malgré une sécrétion initiale de colostrum (1). Un déficit isolé en prolactine (PRL) (taux de PRL à 6,5 ng/ml au 10e jour, nul au 20e jour post-partum) a été identifié dans un contexte d’incapacité à produire du lait malgré une stimulation mammaire et un traitement par dompéridone. Le reste du bilan anté-hypophysaire, l’IRM hypophysaire étaient normaux. Un traitement séquentiel par PRL recombinante par voie sous-cutanée toutes les 12 heures pendant 28 jours a permis de normaliser l’insuffisance lactotrope et d’aboutir à une production de lait dès le 3e jour et d’initier l’allaitement maintenu pendant toute la durée du traitement.
Aucune mutation des régions codantes des gènes de la PRL, du PRF (PRL Releasing Factor) ou son récepteur ainsi que de 7 gènes impliqués dans la différentiation cellulaire lactotrope n’a été identifiée. L’analyse du sérum de la patiente a permis d’identifier, grâce à une technique de double immunofluorescence sur un extrait hypophysaire humain autopsique, la présence d’auto-anticorps dirigés contre un certain groupe de cellules lactotropes. Certaines cellules sécrétrices de PRL n’étaient par reconnues par les auto-anticorps de la patiente et aucune cellule d’autres lignées anté-hypophysaires n’était reconnue.
Devant une insuffisance lactotrope isolée, une étiologie auto-immune peut donc être évoquée, d’autant plus que les patientes présentent un terrain personnel et/ou familial d’auto-immunité.
A l’inverse, Newey et al., viennent de décrire dans le New England Journal of Medicine, une hyperpolactinémie familiale liée à une mutation “perte de fonction” du récepteur de la prolactine (2). Deux sœurs ont présenté une oligoménorrhée et la troisième a consulté pour infertilité. Leurs taux de prolactine étaient compris respectivement entre 95 et 186 ng/ml. Les IRMs hypophysaires étaient normales. Une mutation hétérozygote du récepteur de la prolactine, de transmission a priori autosomique dominante a été identifiée chez les membres de la famille présentant une hyperprolactinémie. Des études in vitro ont confirmé la perte d’activité du récepteur. Ce tableau constitue la première description de résistance à la prolactine dans l’espèce humaine.
Références bibliographiques
1. Iwama S, Welot CK, Romero CJ, Radovick S, Caturegli P. Isolated Prolactin deficiency associated with serum autoantibodies against prolactin-secreting cells. JCEM 2013, 98:3920-5.
2. Newey PJ, Phil D, Gorvin CM et al. Mutant prolactin receptor and familial hyperprolactinemia. New Engl J Med. 2013;369: 2012-20
Dilatation aortique et syndrome de Turner : un nouveau modèle prédictif de l’évolution du diamètre aortique
Bruno Donadille (Paris)
Les patientes avec un syndrome de Turner (ST) sont porteuses de malformations aortiques congénitales, comme une bicuspidie valvulaire aortique (BAV), chez plus de 20 % et/ou une coarctation aortique (CA) chez 12 % des patientes, avec implication récente d’une délétion du bras court du chromosome X (1). La première cause de mortalité des patientes avec un ST est d’origine cardiovasculaire, avec un haut risque de dissection aortique, 100 fois celui de la population générale. S’il est établi que la standardisation du suivi passe désormais par les valeurs du diamètre de l’aorte initiale rapportées à la surface corporelle ou index aortique (N < 20 mm/m²), le suivi prospectif de l’histoire naturelle de cette maladie rare n’avait jusqu’ici fait l’objet que de très peu d’études.
L’équipe de Claus Gravholt (2) vient de décrire le plus long suivi prospectif de leur cohorte monocentrique de 102 patientes adultes avec ST. Les patientes ont été évaluées par une IRM standardisée avec mesure de l’aorte aux 9 niveaux de référence (soit plus de 2 000 valeurs de diamètres aortiques indexés). L’étude comportait 3 visites entre 0 et 5 ans : au départ, à 2,4+/-0,4 ans et à 4,8 +/-0.5 ans (range: 3.5-5.7 ans). Parmi 102 patientes, 78 (76 %) ont terminé l’étude ; elles étaient âgées en moyenne de 38+/- 9,9 ans (range: 19-62 ans), avec une majorité (61 %) de caryotypes 45,X. Seuls les diamètres proximaux de l’aorte se sont majorés. La cinétique de dilatation, est de 0,20 +/- 0,34 mm/an à 0,38 +/-0,46 mm/an. Les variables affectant les diamètres aortiques ont été : la présence de malformation congénitale (BAV, CA, p < 0,0001), l’âge (p < 0,0001), la surface corporelle (p = 0,015), la TA diastolique (p = 0,0008), un traitement anti-HTA (p = 0,005) ; un THS n’étant pas significatif (p = 0,08). Le caryotype 45,X n’a pas été retrouvé significativement prédictif de dilatation, mais il influence indirectement les autres facteurs, comme la présence des malformations elles-mêmes (BAV, CA).
Cette étude produit un modèle mathématique de prédiction du diamètre aortique qui peut être très utile pour améliorer la prise en charge des patientes avec un syndrome de Turner.
Références bibliographiques
1.Bondy C, Bakalov VK, Cheng C, Olivieri L, Rosing DR, Arai AE. Bicuspid aortic valve and aortic coarctation are linked to deletion of the X chromosome short arm in Turner syndrome. J Med Genet. 2013 Oct;50(10):662-5.
2. Mortensen KH, Erlandsen M, Andersen NH, Gravholt CH., Prediction of aortic dilation in Turner syndrome – enhancing the use of serial cardiovascular magnetic resonance. J Cardiovasc Magn Reson, 2013 Jun 6;15(1):47. (Epub ahead of print)